The simulated data simdata_2ct with two cell types is
generated based on the real single-cell data
DuoClustering2018::sce_full_Zhengmix4eq() with the help of
scDesign3. For more details about generating synthetic
data, please check our paper.
The structure of simdata_2ct is as follows:
str(simdata_2ct)
#> List of 4
#> $ simu_sce:Formal class 'SingleCellExperiment' [package "SingleCellExperiment"] with 9 slots
#> .. ..@ int_elementMetadata:Formal class 'DFrame' [package "S4Vectors"] with 6 slots
#> .. .. .. ..@ rownames : NULL
#> .. .. .. ..@ nrows : int 198
#> .. .. .. ..@ listData :List of 1
#> .. .. .. .. ..$ rowPairs:Formal class 'DFrame' [package "S4Vectors"] with 6 slots
#> .. .. .. .. .. .. ..@ rownames : NULL
#> .. .. .. .. .. .. ..@ nrows : int 198
#> .. .. .. .. .. .. ..@ listData : Named list()
#> .. .. .. .. .. .. ..@ elementType : chr "ANY"
#> .. .. .. .. .. .. ..@ elementMetadata: NULL
#> .. .. .. .. .. .. ..@ metadata : list()
#> .. .. .. ..@ elementType : chr "ANY"
#> .. .. .. ..@ elementMetadata: NULL
#> .. .. .. ..@ metadata : list()
#> .. ..@ int_colData :Formal class 'DFrame' [package "S4Vectors"] with 6 slots
#> .. .. .. ..@ rownames : NULL
#> .. .. .. ..@ nrows : int 998
#> .. .. .. ..@ listData :List of 3
#> .. .. .. .. ..$ reducedDims:Formal class 'DFrame' [package "S4Vectors"] with 6 slots
#> .. .. .. .. .. .. ..@ rownames : NULL
#> .. .. .. .. .. .. ..@ nrows : int 998
#> .. .. .. .. .. .. ..@ listData : Named list()
#> .. .. .. .. .. .. ..@ elementType : chr "ANY"
#> .. .. .. .. .. .. ..@ elementMetadata: NULL
#> .. .. .. .. .. .. ..@ metadata : list()
#> .. .. .. .. ..$ altExps :Formal class 'DFrame' [package "S4Vectors"] with 6 slots
#> .. .. .. .. .. .. ..@ rownames : NULL
#> .. .. .. .. .. .. ..@ nrows : int 998
#> .. .. .. .. .. .. ..@ listData : Named list()
#> .. .. .. .. .. .. ..@ elementType : chr "ANY"
#> .. .. .. .. .. .. ..@ elementMetadata: NULL
#> .. .. .. .. .. .. ..@ metadata : list()
#> .. .. .. .. ..$ colPairs :Formal class 'DFrame' [package "S4Vectors"] with 6 slots
#> .. .. .. .. .. .. ..@ rownames : NULL
#> .. .. .. .. .. .. ..@ nrows : int 998
#> .. .. .. .. .. .. ..@ listData : Named list()
#> .. .. .. .. .. .. ..@ elementType : chr "ANY"
#> .. .. .. .. .. .. ..@ elementMetadata: NULL
#> .. .. .. .. .. .. ..@ metadata : list()
#> .. .. .. ..@ elementType : chr "ANY"
#> .. .. .. ..@ elementMetadata: NULL
#> .. .. .. ..@ metadata : list()
#> .. ..@ int_metadata :List of 1
#> .. .. ..$ version:Classes 'package_version', 'numeric_version' hidden list of 1
#> .. .. .. ..$ : int [1:3] 1 24 0
#> .. ..@ rowRanges :Formal class 'CompressedGRangesList' [package "GenomicRanges"] with 5 slots
#> .. .. .. ..@ unlistData :Formal class 'GRanges' [package "GenomicRanges"] with 7 slots
#> .. .. .. .. .. ..@ seqnames :Formal class 'Rle' [package "S4Vectors"] with 4 slots
#> .. .. .. .. .. .. .. ..@ values : Factor w/ 0 levels:
#> .. .. .. .. .. .. .. ..@ lengths : int(0)
#> .. .. .. .. .. .. .. ..@ elementMetadata: NULL
#> .. .. .. .. .. .. .. ..@ metadata : list()
#> .. .. .. .. .. ..@ ranges :Formal class 'IRanges' [package "IRanges"] with 6 slots
#> .. .. .. .. .. .. .. ..@ start : int(0)
#> .. .. .. .. .. .. .. ..@ width : int(0)
#> .. .. .. .. .. .. .. ..@ NAMES : NULL
#> .. .. .. .. .. .. .. ..@ elementType : chr "ANY"
#> .. .. .. .. .. .. .. ..@ elementMetadata: NULL
#> .. .. .. .. .. .. .. ..@ metadata : list()
#> .. .. .. .. .. ..@ strand :Formal class 'Rle' [package "S4Vectors"] with 4 slots
#> .. .. .. .. .. .. .. ..@ values : Factor w/ 3 levels "+","-","*":
#> .. .. .. .. .. .. .. ..@ lengths : int(0)
#> .. .. .. .. .. .. .. ..@ elementMetadata: NULL
#> .. .. .. .. .. .. .. ..@ metadata : list()
#> .. .. .. .. .. ..@ seqinfo :Formal class 'Seqinfo' [package "GenomeInfoDb"] with 4 slots
#> .. .. .. .. .. .. .. ..@ seqnames : chr(0)
#> .. .. .. .. .. .. .. ..@ seqlengths : int(0)
#> .. .. .. .. .. .. .. ..@ is_circular: logi(0)
#> .. .. .. .. .. .. .. ..@ genome : chr(0)
#> .. .. .. .. .. ..@ elementMetadata:Formal class 'DFrame' [package "S4Vectors"] with 6 slots
#> .. .. .. .. .. .. .. ..@ rownames : NULL
#> .. .. .. .. .. .. .. ..@ nrows : int 0
#> .. .. .. .. .. .. .. ..@ listData : Named list()
#> .. .. .. .. .. .. .. ..@ elementType : chr "ANY"
#> .. .. .. .. .. .. .. ..@ elementMetadata: NULL
#> .. .. .. .. .. .. .. ..@ metadata : list()
#> .. .. .. .. .. ..@ elementType : chr "ANY"
#> .. .. .. .. .. ..@ metadata : list()
#> .. .. .. ..@ elementMetadata:Formal class 'DFrame' [package "S4Vectors"] with 6 slots
#> .. .. .. .. .. ..@ rownames : NULL
#> .. .. .. .. .. ..@ nrows : int 198
#> .. .. .. .. .. ..@ listData : Named list()
#> .. .. .. .. .. ..@ elementType : chr "ANY"
#> .. .. .. .. .. ..@ elementMetadata: NULL
#> .. .. .. .. .. ..@ metadata : list()
#> .. .. .. ..@ elementType : chr "GRanges"
#> .. .. .. ..@ metadata : list()
#> .. .. .. ..@ partitioning :Formal class 'PartitioningByEnd' [package "IRanges"] with 5 slots
#> .. .. .. .. .. ..@ end : int [1:198] 0 0 0 0 0 0 0 0 0 0 ...
#> .. .. .. .. .. ..@ NAMES : chr [1:198] "ENSG00000116251" "ENSG00000142676" "ENSG00000142669" "ENSG00000169442" ...
#> .. .. .. .. .. ..@ elementType : chr "ANY"
#> .. .. .. .. .. ..@ elementMetadata: NULL
#> .. .. .. .. .. ..@ metadata : list()
#> .. ..@ colData :Formal class 'DFrame' [package "S4Vectors"] with 6 slots
#> .. .. .. ..@ rownames : chr [1:998] "naive.cytotoxic10013" "naive.cytotoxic5827" "naive.cytotoxic1319" "naive.cytotoxic4199" ...
#> .. .. .. ..@ nrows : int 998
#> .. .. .. ..@ listData :List of 1
#> .. .. .. .. ..$ cell_type: chr [1:998] "celltype0" "celltype0" "celltype0" "celltype0" ...
#> .. .. .. ..@ elementType : chr "ANY"
#> .. .. .. ..@ elementMetadata: NULL
#> .. .. .. ..@ metadata : list()
#> .. ..@ assays :Formal class 'SimpleAssays' [package "SummarizedExperiment"] with 1 slot
#> .. .. .. ..@ data:Formal class 'SimpleList' [package "S4Vectors"] with 4 slots
#> .. .. .. .. .. ..@ listData :List of 2
#> .. .. .. .. .. .. ..$ counts : num [1:198, 1:998] 3 70 2 3 1 7 0 6 0 1 ...
#> .. .. .. .. .. .. .. ..- attr(*, "dimnames")=List of 2
#> .. .. .. .. .. .. .. .. ..$ : chr [1:198] "ENSG00000116251" "ENSG00000142676" "ENSG00000142669" "ENSG00000169442" ...
#> .. .. .. .. .. .. .. .. ..$ : chr [1:998] "naive.cytotoxic10013" "naive.cytotoxic5827" "naive.cytotoxic1319" "naive.cytotoxic4199" ...
#> .. .. .. .. .. .. ..$ logcounts: num [1:198, 1:998] 1.386 4.263 1.099 1.386 0.693 ...
#> .. .. .. .. .. .. .. ..- attr(*, "dimnames")=List of 2
#> .. .. .. .. .. .. .. .. ..$ : chr [1:198] "ENSG00000116251" "ENSG00000142676" "ENSG00000142669" "ENSG00000169442" ...
#> .. .. .. .. .. .. .. .. ..$ : chr [1:998] "naive.cytotoxic10013" "naive.cytotoxic5827" "naive.cytotoxic1319" "naive.cytotoxic4199" ...
#> .. .. .. .. .. ..@ elementType : chr "ANY"
#> .. .. .. .. .. ..@ elementMetadata: NULL
#> .. .. .. .. .. ..@ metadata : list()
#> .. ..@ NAMES : NULL
#> .. ..@ elementMetadata :Formal class 'DFrame' [package "S4Vectors"] with 6 slots
#> .. .. .. ..@ rownames : NULL
#> .. .. .. ..@ nrows : int 198
#> .. .. .. ..@ listData : Named list()
#> .. .. .. ..@ elementType : chr "ANY"
#> .. .. .. ..@ elementMetadata: NULL
#> .. .. .. ..@ metadata : list()
#> .. ..@ metadata : list()
#> $ de_idx : Named int [1:20] 2 15 16 24 34 35 55 64 71 86 ...
#> ..- attr(*, "names")= chr [1:20] "ENSG00000142676" "ENSG00000143947" "ENSG00000034510" "ENSG00000144713" ...
#> $ ct0_idx : int [1:499] 1 2 3 4 5 6 7 8 9 10 ...
#> $ ct1_idx : int [1:499] 500 501 502 503 504 505 506 507 508 509 ...The true differentially expressed (DE) genes are
truth = names(simdata_2ct$de_idx)
truth
#> [1] "ENSG00000142676" "ENSG00000143947" "ENSG00000034510" "ENSG00000144713"
#> [5] "ENSG00000109475" "ENSG00000145425" "ENSG00000112306" "ENSG00000205542"
#> [9] "ENSG00000147604" "ENSG00000177600" "ENSG00000166441" "ENSG00000251562"
#> [13] "ENSG00000122026" "ENSG00000167526" "ENSG00000198242" "ENSG00000108298"
#> [17] "ENSG00000115268" "ENSG00000105640" "ENSG00000105372" "ENSG00000083845"Step 1: Perform Testing-after-Clustering on Each Half via (Multiple) Data Splitting
mss = mds1(simdata_2ct$simu_sce,
M = 1, # times of data splitting
params1 = list(normalized_method = "none"), # parameters for the first half
params2 = list(normalized_method = "none")) # parameters for the second half
#>
#> ===== Multiple Data Splitting: 1 / 1 =====
#>
#> ----- data splitting (1st half) -----
#>
#> ----- data splitting (2nd half) ----It returns the mirror statistics,
hist(mss[[1]], breaks = 50)
cutoff = calc_tau(mss[[1]], q = 0.05)
abline(v = cutoff, lwd = 2, col = "red")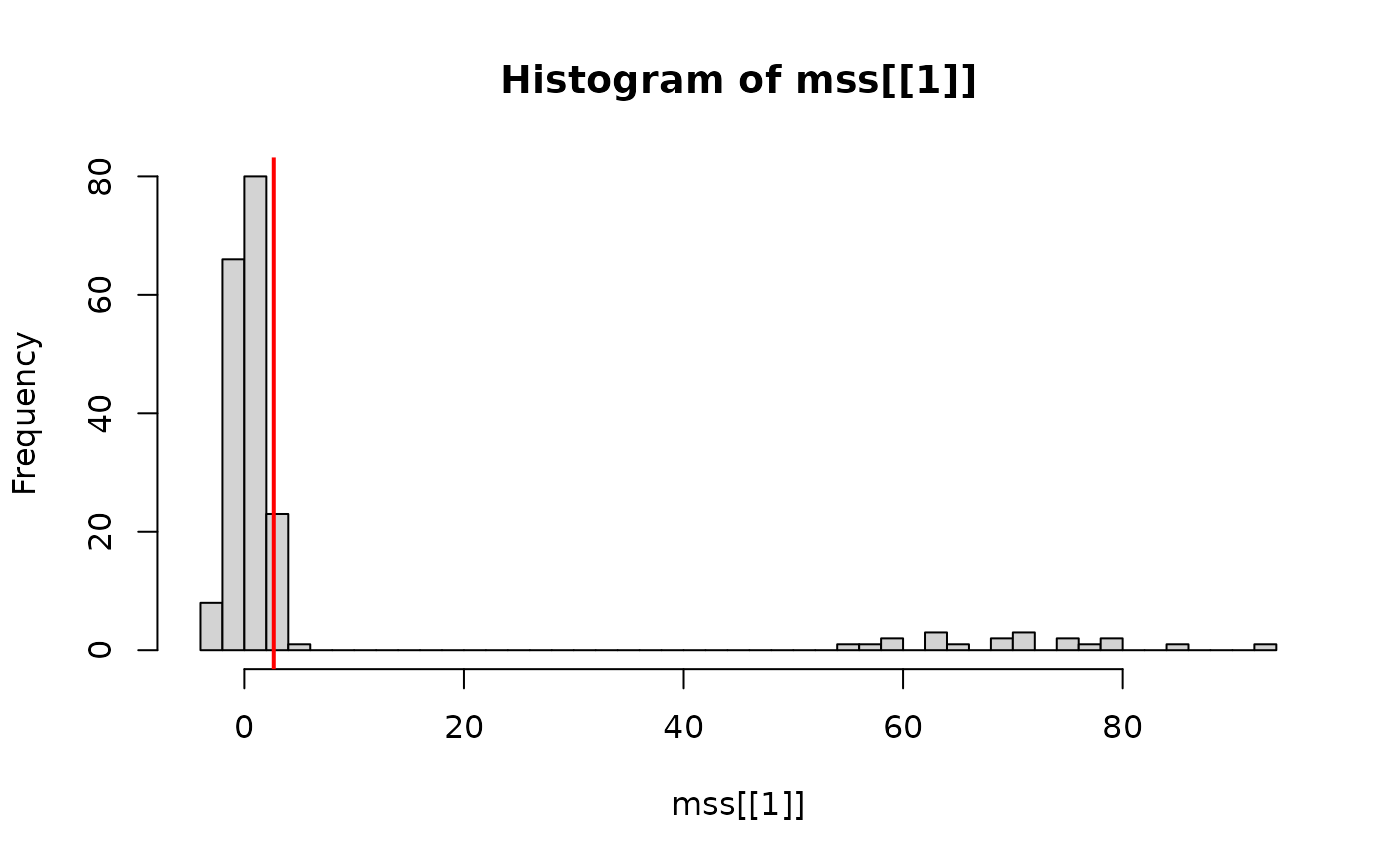
In this step, cutoff is chosen to control the nominal
False Discovery Rate (FDR) at q = 0.05.
Note that the above histogram is for the results from the first data
splitting mss[[1]]. If with multiple data splitting, we can
aggregate the results by calculating the inclusion rate for each gene,
and also calculate a cutoff. Then, either for DS or MDS, we can obtain
the estimation of the significant DE gene set. We wrap this as the
second step.
For multiple data splitting (MDS), we can aggregate results by calculating the inclusion rate for each gene and determining a new cutoff. This allows us to estimate the set of significant DE genes, whether using DS or MDS. We wrap this process as the second step:
Step 2. Calculate Cutoff and Estimate the Selection Set
sel = mds2(mss, q = 0.05)
#> use the tie.method = fair
sel
#> [1] "ENSG00000144713" "ENSG00000167526" "ENSG00000115268" "ENSG00000105640"
#> [5] "ENSG00000108298" "ENSG00000177600" "ENSG00000198242" "ENSG00000112306"
#> [9] "ENSG00000142676" "ENSG00000145425" "ENSG00000105372" "ENSG00000083845"
#> [13] "ENSG00000166441" "ENSG00000122026" "ENSG00000143947" "ENSG00000205542"
#> [17] "ENSG00000034510" "ENSG00000109475" "ENSG00000251562" "ENSG00000147604"
#> [21] "ENSG00000138326" "ENSG00000140264" "ENSG00000102879" "ENSG00000116251"
#> [25] "ENSG00000185201" "ENSG00000140319" "ENSG00000148908"we can also evaluate the accuracy
calc_acc(sel, truth)
#> fdr power f1
#> 0.2592593 1.0000000 0.8510638A Whole Step
As a whole, the above two steps mds1 and
mds2 have been wrapped into a single function
mds, and you can simply run the following command to obtain
the selection set.
sel = mds(simdata_2ct$simu_sce,
q = 0.05,
M = 1,
params1 = list(normalized_method = "none"),
params2 = list(normalized_method = "none"))
#>
#> ===== Multiple Data Splitting: 1 / 1 =====
#>
#> ----- data splitting (1st half) -----
#>
#> ----- data splitting (2nd half) ----
#> use the tie.method = fairAs a comparison, we also check the naive double-dipping method.
sel.dd = dd(simdata_2ct$simu_sce, params = list(normalized_method = "none"), q = 0.05, test.use = "t")and the accuracy is
calc_acc(sel.dd, truth)
#> fdr power f1
#> 0 1 1Normalization
In practice, to account for the library size, a normalization method is needed. One suitable approach is the SCT transform.
sel.sct = mds(simdata_2ct$simu_sce,
q = 0.05,
M = 1,
params1 = list(normalized_method = "sct"),
params2 = list(normalized_method = "sct"))
#>
#> ===== Multiple Data Splitting: 1 / 1 =====
#>
#> ----- data splitting (1st half) -----
#>
#> ----- data splitting (2nd half) ----
#> use the tie.method = fair
sel.dd.sct = dd(simdata_2ct$simu_sce, params = list(normalized_method = "sct"))
#> Warning: The following arguments are not used: norm.method
sapply(list(ds = sel.sct, dd = sel.dd.sct), function(x) calc_acc(x, names(simdata_2ct$de_idx)))
#> ds dd
#> fdr 0.1304348 0.8245614
#> power 1.0000000 1.0000000
#> f1 0.9302326 0.2985075Whitening
We recommend the whitening strategy for weak signal and (highly) correlated data.
sel = mds(simdata_2ct$simu_sce,
q = 0.05,
M = 1,
params1 = list(normalized_method = "sct", pca.whiten = TRUE),
params2 = list(normalized_method = "sct", pca.whiten = TRUE))
#>
#> ===== Multiple Data Splitting: 1 / 1 =====
#>
#> ----- data splitting (1st half) -----
#>
#> ----- data splitting (2nd half) ----
#> use the tie.method = fair
sel.dd = dd(simdata_2ct$simu_sce, params = list(normalized_method = "sct", pca.whiten = TRUE))
#> Warning: The following arguments are not used: norm.method
sapply(list(ds = sel, dd = sel.dd), function(x) calc_acc(x, names(simdata_2ct$de_idx)))
#> ds dd
#> fdr 0.09090909 0.8290598
#> power 1.00000000 1.0000000
#> f1 0.95238095 0.2919708Session Info
sessionInfo()
#> R version 4.1.3 (2022-03-10)
#> Platform: x86_64-pc-linux-gnu (64-bit)
#> Running under: Ubuntu 24.04.3 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so
#>
#> locale:
#> [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
#> [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
#> [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
#> [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
#>
#> attached base packages:
#> [1] stats graphics grDevices datasets utils methods base
#>
#> other attached packages:
#> [1] future_1.69.0 SplitClusterTest_0.1.4
#>
#> loaded via a namespace (and not attached):
#> [1] copula_1.1-6 spatstat.univar_3.0-1
#> [3] spam_2.11-0 systemfonts_1.1.0
#> [5] plyr_1.8.9 igraph_2.1.1
#> [7] lazyeval_0.2.2 sp_2.1-4
#> [9] splines_4.1.3 pspline_1.0-21
#> [11] listenv_0.9.1 scattermore_1.2
#> [13] GenomeInfoDb_1.30.1 ggplot2_3.5.1
#> [15] digest_0.6.37 htmltools_0.5.8.1
#> [17] fansi_1.0.6 magrittr_2.0.3
#> [19] tensor_1.5 cluster_2.1.2
#> [21] ROCR_1.0-11 globals_0.18.0
#> [23] matrixStats_1.4.1 stabledist_0.7-2
#> [25] pkgdown_2.2.0 spatstat.sparse_3.1-0
#> [27] colorspace_2.1-1 ggrepel_0.9.6
#> [29] textshaping_0.4.0 xfun_0.56
#> [31] dplyr_1.1.4 RCurl_1.98-1.16
#> [33] jsonlite_1.8.9 progressr_0.14.0
#> [35] spatstat.data_3.1-2 survival_3.2-13
#> [37] zoo_1.8-12 glue_1.8.0
#> [39] polyclip_1.10-7 gtable_0.3.5
#> [41] zlibbioc_1.40.0 XVector_0.34.0
#> [43] leiden_0.4.3.1 DelayedArray_0.20.0
#> [45] future.apply_1.20.1 SingleCellExperiment_1.16.0
#> [47] BiocGenerics_0.40.0 abind_1.4-8
#> [49] scales_1.3.0 mvtnorm_1.3-3
#> [51] spatstat.random_3.3-2 miniUI_0.1.1.1
#> [53] Rcpp_1.0.13 viridisLite_0.4.2
#> [55] xtable_1.8-4 reticulate_1.39.0
#> [57] dotCall64_1.2 stats4_4.1.3
#> [59] htmlwidgets_1.6.4 httr_1.4.7
#> [61] RColorBrewer_1.1-3 Seurat_4.4.0
#> [63] ica_1.0-3 pkgconfig_2.0.3
#> [65] farver_2.1.2 sass_0.4.9
#> [67] uwot_0.2.2 deldir_2.0-4
#> [69] utf8_1.2.4 tidyselect_1.2.1
#> [71] rlang_1.1.4 reshape2_1.4.4
#> [73] later_1.3.2 munsell_0.5.1
#> [75] tools_4.1.3 cachem_1.1.0
#> [77] cli_3.6.3 generics_0.1.3
#> [79] ggridges_0.5.6 evaluate_1.0.1
#> [81] stringr_1.5.1 fastmap_1.2.0
#> [83] yaml_2.3.10 ragg_1.5.0
#> [85] goftest_1.2-3 knitr_1.51
#> [87] fs_1.6.4 fitdistrplus_1.2-1
#> [89] purrr_1.0.2 RANN_2.6.2
#> [91] pbapply_1.7-4 nlme_3.1-155
#> [93] mime_0.12 compiler_4.1.3
#> [95] plotly_4.10.4 png_0.1-8
#> [97] spatstat.utils_3.1-0 tibble_3.2.1
#> [99] pcaPP_2.0-5 gsl_2.1-7
#> [101] bslib_0.8.0 stringi_1.8.4
#> [103] desc_1.4.3 lattice_0.20-45
#> [105] Matrix_1.6-5 vctrs_0.6.5
#> [107] pillar_1.9.0 lifecycle_1.0.4
#> [109] ADGofTest_0.3 BiocManager_1.30.25
#> [111] spatstat.geom_3.3-3 lmtest_0.9-40
#> [113] jquerylib_0.1.4 RcppAnnoy_0.0.22
#> [115] data.table_1.16.2 cowplot_1.1.3
#> [117] bitops_1.0-9 irlba_2.3.5.1
#> [119] httpuv_1.6.15 patchwork_1.3.0
#> [121] GenomicRanges_1.46.1 R6_2.5.1
#> [123] promises_1.3.0 renv_1.0.10
#> [125] KernSmooth_2.23-20 gridExtra_2.3
#> [127] IRanges_2.28.0 parallelly_1.46.1
#> [129] codetools_0.2-18 MASS_7.3-55
#> [131] SummarizedExperiment_1.24.0 SeuratObject_5.0.2
#> [133] sctransform_0.4.1 S4Vectors_0.32.4
#> [135] GenomeInfoDbData_1.2.7 parallel_4.1.3
#> [137] grid_4.1.3 tidyr_1.3.1
#> [139] rmarkdown_2.30 MatrixGenerics_1.6.0
#> [141] Rtsne_0.17 spatstat.explore_3.3-3
#> [143] numDeriv_2016.8-1.1 Biobase_2.54.0
#> [145] shiny_1.9.1